| layout | subdomain |
|---|---|
subsite-galaxy |
graphclust |
Welcome to GraphClust2 Galaxy server for clustering of RNAs according to sequence and secondary structures similarities.
GraphClust2 is a workflow for scalable clustering of RNAs based on sequence and secondary structures feature. GraphClust2 is implemented within the Galaxy framework and consists a set of integrated Galaxy tools and flavors of the linear-time clustering workflow.
- TOC {:toc}
GraphClust2 rapid start
Interactive tours are available for Galaxy and GraphClust2. To run the tours please on top panel go to Help→Interactive Tours and click on one of the tours prefixed GraphClust workflow (direct link to the basic tour{:target="_blank"}). Please use your personal user-password for logging in. You can check the other tours for a more general introduction to the Galaxy interface.
Galaxy interface
Are you new to Galaxy, or returning after a long time, and looking for help to get started? Take a guided tour{:target="_blank"} through Galaxy's user interface.
Please also refer to the GraphClust2 repository{:target="_blank"} for other deployment options and manuals.
This video tutorial{:target="_blank"} can be helpful to get a visually comprehensive introduction on setting-up and running GraphClust2. The video starts with setting up the docker Galaxy server that can be skipped through using this server.
 {:target="_blank"}
{:target="_blank"}
A comprehensive set of pre-configured flavors of GraphClust2 are provided and described inside the workflows directory{:target="_blank"} There you can find the alternative pre-configurations of GraphClust-2 as flavors tailored for different use-case scenarios.
Below workflows can be directly accessed on the public server. For the extended description and alternatives please refer to the github workflows directory{:target="_blank"}
-
The MotifFinder workflow flavor targets identifying a handful of local signals/motifs under the likely presence of noise and sequence context.
- MotifFinder: GraphClust-MotifFinder{:target="_blank"}
-
The pre-configured main workflows perform best for clustering and partitioning a set of RNA sequences with quasi defined structure boundary signals (e.g. ncRNAs or data from genomic screenings with tools such as CMfinder or RNAz screens). Usually one to three rounds of clustering would be enough for typical scenarios. You may find further suitable pre-configured flavors from the github directory page.
- Workflow main: GraphClust_1r{:target="_blank"}
- Workflow main, pre-configured for two rounds : GraphClust_2r{:target="_blank"}
To import or upload additional workflow flavors (e.g. from workflows directory), on the top panel go to Workflow menu. On top right side of the screen click on "Upload or import workflow" button. You can either upload workflow from your local system or by providing the URL of the workflow. Log in is necessary to access into the workflow menu. The docker galaxy instance has a pre-configured easy! info that can be found by following the interactive tour. You can download workflows from the following links
The histories shared and linked below, corresponds to the clustering analysis and evaluations that are performed and presented in the GraphClust2 paper.
- NEAT1 clustering history{:target="_blank"}
- MALAT1 clustering history{:target="_blank"}
- HOTAIR clustering history{:target="_blank"}
- XIST clustering history{:target="_blank"}
- FTL clustering history{:target="_blank"}
- MAF to fasta conversions{:target="_blank"}
- SLBP clustering history{:target="_blank"}
- SLBP RF00032 seed (mixed with 98.5% background) clustering history{:target="_blank"}
- Roquin1{:target="_blank"}
- Marine metatranscriptome clustering history{:target="_blank"}
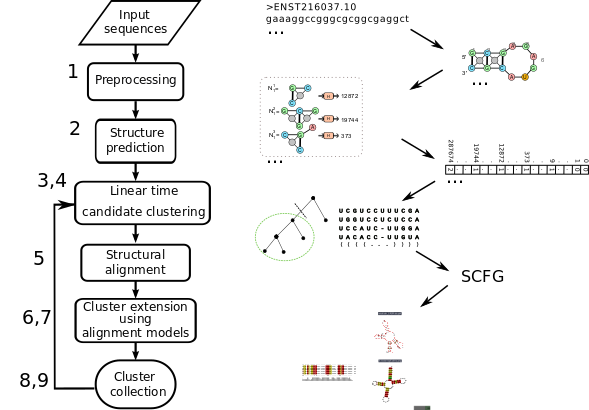
The pipeline for clustering RNA sequences and structured motif discovery is a multi-step pipeline. Overall it consists of three major phases: a) sequence based pre-clustering b) encoding predicted RNA structures as graph features c) iterative fast candidate clustering then refinement

 {:target="_blank"}
{:target="_blank"}
Below is a coarse-grained correspondence list of GraphClust2 tool names with each step:
| Stage | Galaxy Tool Name | Description |
|---|---|---|
| 1 | {% include tool.html id="graphclust_preprocessing" label="Graphclust Preprocessing" %} | Input preprocessing (fragmentation) |
| 2 | {% include tool.html id="fasta_to_gspan" %} | Generation of structures via RNAshapes and conversion into graphs |
| 3 | {% include tool.html id="NSPDK_sparseVect" %} | Generation of graph features via NSPDK |
| 4 | {% include tool.html id="NSPDK_candidateClusters" %} | min-hash based clustering of all feature vectors, output top dense candidate clusters |
| 5 | {% include tool.html id="PGMA_locarna" %} | Locarna based clustering of each candidate cluster, all-vs-all pairwise alignments, create multiple alignments along guide tree, select best subtree, and refine alignment. |
| 6 | {% include tool.html id="infernal_cmbuild" label="Build covariance models" %} | create candidate model |
| 7 | {% include tool.html id="infernal_cmsearch" label="Search covariance models" %} | Scan full input sequences with Infernal's cmsearch to find missing cluster members |
| 8,9 | {% include tool.html id="graphclust_postprocessing" label="Report Results" %} and {% include tool.html id="graphclust_aggregate_alignments" label="conservation evaluations" %} | Collect final clusters and create example alignments of top cluster members |
| {: .table.table-striped} |
The input to the workflow is a set of putative RNA sequences in FASTA format. Inside the data directory within the repository, you can find examples of the input format.
The output contains the predicted clusters, where similar putative input RNA sequences form a cluster. Additionally overall status of the clusters and the matching of cluster elements is reported for each cluster.
Please proceed with the interactive tour named GraphClust workflow step by step, available under Help->Interactive Tours
Please refer to the in-wrapper help descriptions the tools documentations and the repository's FAQs{:target="_blank"} for checking the important parameters.
You can file an github issue{:target="_blank"} or find our contact information in the Backofen lab page{:target="_blank"}.
- Miladi, Milad, Eteri Sokhoyan, Torsten Houwaart, Steffen Heyne, Fabrizio Costa, Bjoern Gruening, and Rolf Backofen. "GraphClust2: annotation and discovery of structured RNAs with scalable and accessible integrative clustering" bioRxiv (2019): 550335. doi: https://doi.org/10.1101/550335{:target="_blank"}
- Milad Miladi, Björn Grüning, & Eteri Sokhoyan. BackofenLab/GraphClust-2: Zenodo. http://doi.org/10.5281/zenodo.1135094