anvi'o v4, "rosalind"
We are happy to announce a new version of anvi'o, "rosalind".
After nearly 300 changes that introduced about 15,000 new lines, and removed about 7,500 from the anvi'o codebase, the current version includes many bug fixes, as well as some new features. This release note intends to give you a summary of most important changes.

The codename is a small tribute to Rosalind Franklin, the British biophysicist whose work, among other advances in life sciences, led to the discovery of the DNA double helix. This codename was inspired by Emily Crossette's suggestion, 'esther', "after Esther Lederberg, who co-developed a replica plating method with her husband but was largely unrecognized and discriminated against as a woman scientist". Emily explained that her suggestion was to "celebrate how far we have come as a scientific community and look to the future". Yes. We fortunately did not stay where we were, but we are still far from where we could have been. We remember these women and many others with respect and gratitude, and understand our responsibility to make sure the younger generations of scientists will not suffer from the kinds of discrimination to which their professors were subjected.
An elegant way to upgrade anvi'o databases
Upgrading anvi'o databases is now simpler than ever. With this change, the number of excuses you can use to not switch to the newest version of anvi'o goes from "0" to "-1". Just saying.
We now have a single program, anvi-migrate-db, that upgrades any anvi'o database to the latest version in one step.
As a part of this change we replaced all HDF5 files, which resulted in tremendous performance gain (especially in pangenomic operations that required access to the genome storage database), and up to 10-fold reduction in disk storage needs (for auxiliary data files). As a result, these changes did occur: No more CONTIGS.h5 --the content of this file is now a part of the CONTIGS.db (yay for less clutter). No more SAMPLES.db (more on this down below). Genome storage and auxiliary data files now have .db extensions rather than .h5 as they are now SQLite databases, instead of HD5 files.
Improvements in the pangenomic workflow
We made multiple very critical improvements in our pangenomic workflow. Here is a list of them:
These are the gene clusters you are looking for. Now it is possible to "select" gene clusters programmatically both from the command line, and from the anvi'o interactive interface through the combination of filters. We thank Ryan Bartelme for pushing us to improve our pangenomic workflow as he once again did in #668. Gene clusters that match to these filters are highlighted immediately on the interface, and can be added into any bin/collection for summary:

Search gene clusters by function. We also now have the capacity to search for gene clusters that describe genes with functions of interest through the command line as well as the interactive interface:
Parallel alignment. After identifying all gene clusters in a given pangenome, anvi'o by default would use muscle or famsa to store multiple sequence alignments for amino acid sequences in each gene cluster. This was one of the most time consuming steps of the pangenomic workflow. With v4, anvi'o uses as many cores as you wish anvi'o to use to parallelize amino acid alignments per gene cluster. It changes a lot.
Forced synteny. Gene clusters in a pangenome are by default organized based on their distribution across genomes (so that is the dendrogram in the center). However, with this version there are additional ways to order them, including ordering them by "synteny". In this forced organization you get to choose one of the genomes in your analysis from the "item orders" combo box, which tells anvi'o that you wish to order all gene clusters in your pangenome based on the order of genes in that genome. We found it to be an efficient way to study missing genomic loci, and other not-so-straightforward-to-spot phenomena.
Everything is better in color. Arguably, one of the most important improvements to the pangenomic workflow was the addition of an amino acid alignment conservancy coloring algorithm. This was done in #732 by Mahmoud Yousef, who is currently a second year Computer Science student at the University of Chicago. Mahmoud also very kindly wrote a blog post to explain the details of this algorithm with examples: http://merenlab.org/2018/02/13/color-coding-aa-alignments/.
Gene popups. Now you can click gene caller ids next to the amino acid sequence alignment in inspection pages, and enjoy these functional popups to access any information (#680):

Cleaner terminology. After consulting with the community, we changed all instances of 'protein clusters' in our pangenomic workflow with 'gene clusters'.
Metapangenomics: linking pangenomes and metagenomes
Anvi'o comes with powerful analytical tools to study pangenomes and metagenomes. Now you can take things one step further with the same ease-of-use.
We define metapangenomics as the outcome of the analysis of pangenomes in conjunction with the environment where the abundance and prevalence of gene clusters and genomes are recovered through shotgun metagenomes. This version includes a new program, anvi-meta-pan-genome, that brings the power of metapangenomics into a single command line. Please read our paper on the Prochlorococcus metapangenome to see how this concept could apply to your research.
Improvements in the interactive interface
This release also include multiple notable improvements in the interactive interface.
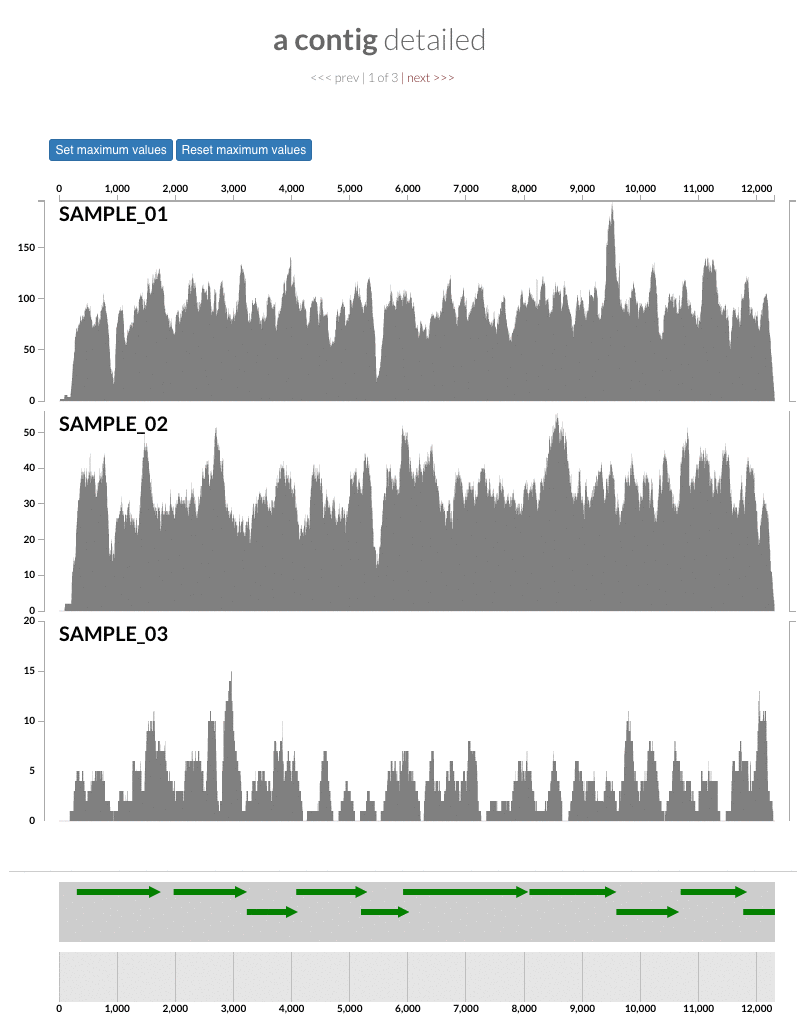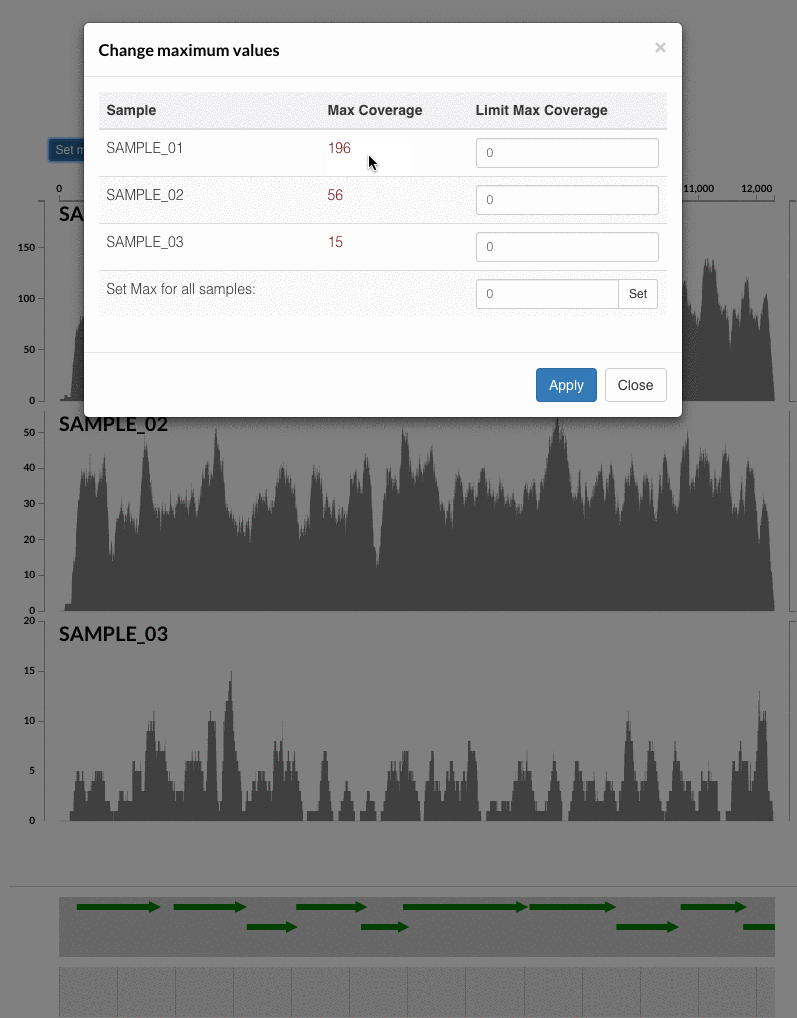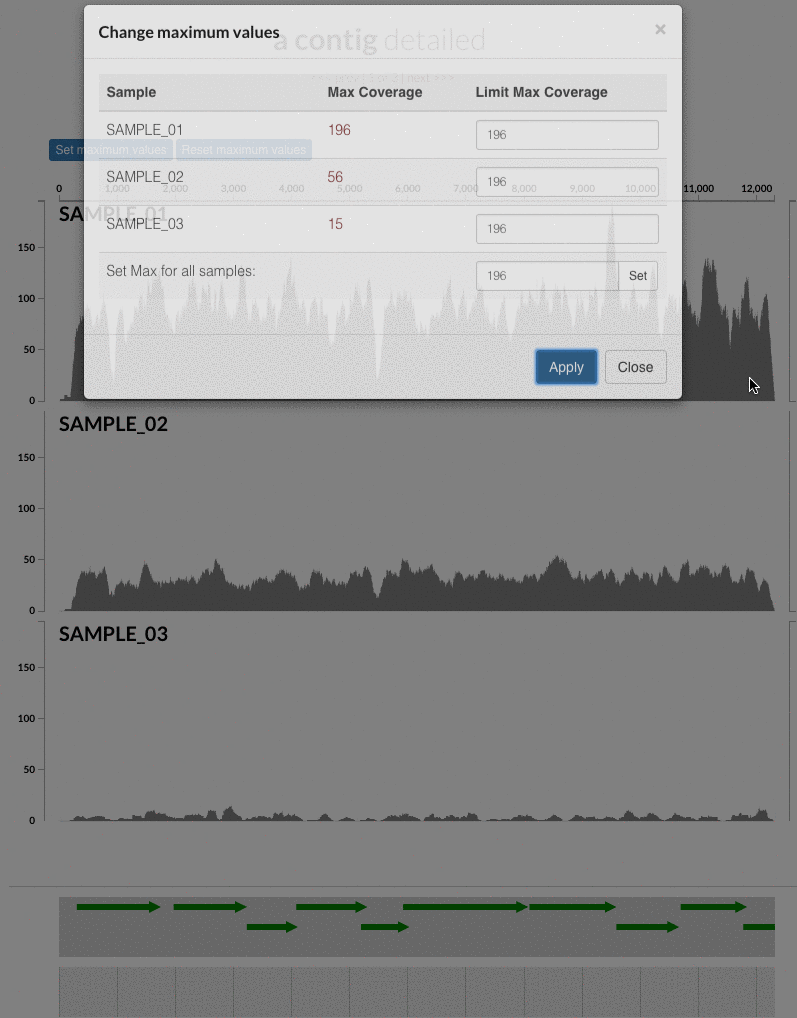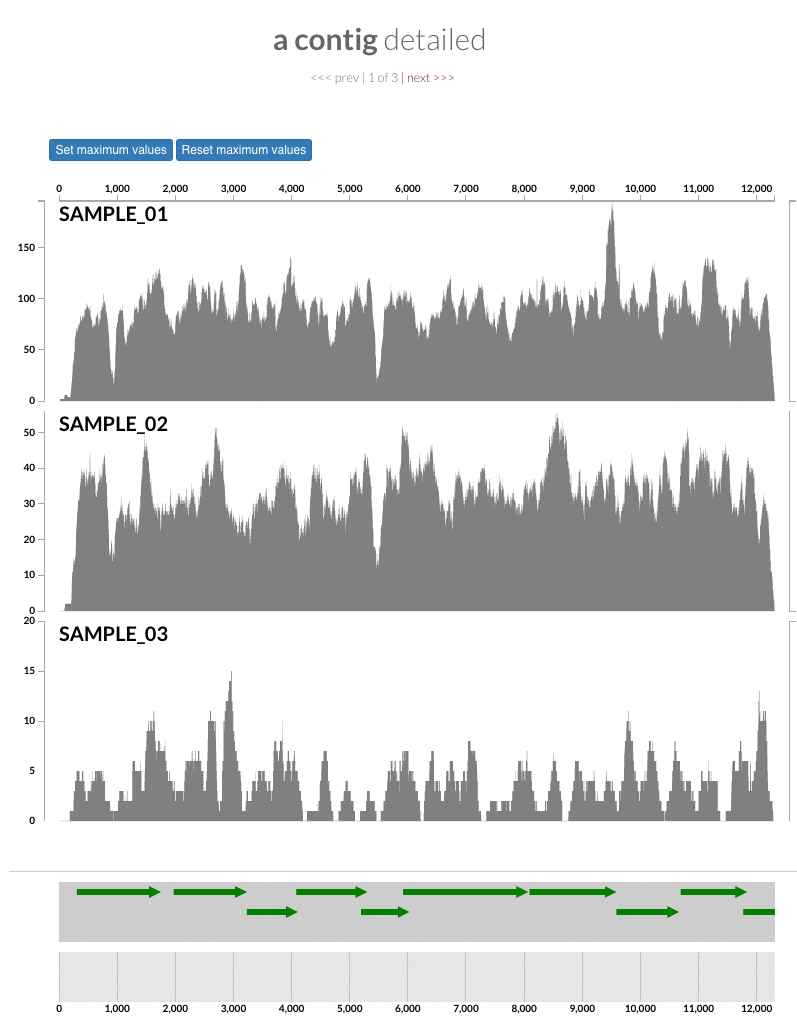
The 'max coverage' fix we all needed but didn't know. Inspection pages are great to investigate coverage data and single-nucleotide variants in a single-nucleotide resolution, however, it was not quite easy to make visual sense of data when coverage values dramatically differed between samples, or short but non-specific mapping pushed maximum coverage values too high to make sense of the actual population coverage in the context of long contigs. In v4 you will see additional buttons in the inspection pages to mitigate these kinds of visual imperfections. Here is some action for you skeptics:
Descriptions tab gets 1-up. One of the most useful features of the interactive interface is the "Descriptions" tab. Yes, we know you are not using it, but you should. Here is an example to see why you should use them (just wait until the page loads, and see all the information that will show up in the right panel): https://anvi-server.org/merenlab/dwh_o_desum. The description tab is extremely useful to take notes and store them in a profile database to remember later. With this new version, you will be able to point out to an item (#715), which will give access to the reader so they can see where it is on the display by highlighting it, or they can inspect it by clicking 'inspect':

Gene mode: a new, highly-resolved interactive mode to study genome bins
This is yet another way for you to examine your data in high-resolution.
We added a flag to anvi-interactive: --gene-mode. When you use this flag along with a collection and a bin name, it allows you to load the interactive interface in the "gene mode". In this mode every item is a gene, instead of a contig, and you can see the coverage, detection, non-outlier coverage, and non-outlier standard deviation of coverage statistics per gene, independently. You can use these data to order the genes, and order the samples. Inspection of nucleotide level coverages, gene sequences, and even gene functions could also be explored in this mode. This allowed us to easily recognize genes that recruit a lot of non-specific mapping, and identify hyper-variable regions in our genomes. One can also search for genes with certain functions, and see their coverages, and the coverage of the genes that are next to them.
Please refer to the help menu for the interactive interface (via anvi-interactive -h or here) to find out more about this mode.
We are excited about this new feature, and we plan to expand it in future versions of anvi'o. If you have any suggestions/complaints/compliments please leave a comment in this issue: #754. We will soon put a tutorial for this mode online, so stay tuned!
A new and elegant way to extend anvi'o displays: additional data tables
We made a major change in our design to simplify the way various data for items and layers can be imported into anvi'o profile or pan databases, and managed. This change opens doors for endless possibilities to manipulate additional data streams through interactive, command line, and application programmer interfaces. Please see a detailed description on this new framework here: http://merenlab.org/2017/12/11/additional-data-tables/.
Other improvements
Optional noise cutoffs for HMMs. This has been a long standing issue (detailed in #498). The current version allows user defined noisee-cutoff terms and make it easier for anyone who wish to make anvi'o use their own HMM collection.
Anvi'o vignettes. All anvi'o programs, their categories, parameters, and help: http://merenlab.org/software/anvio/vignette/. This is big, guys.
Variability performance improvement. Anvi'o now relies on pandas (not the animal, sadly, but the library) to take care of variability operations. While this will not impact user experience much, the code is much more elegant now and we wanted you to know it. See the Pull Request at #660 for more details.
Anvi'o disco mode [ON]. We heard more than once that people do not realize that they need to click the 'Draw' button in the interface if there is no default state to load and draw everything automtically (#739). So, disco:

New scripts and programs
We have some new programs that comes with this version. Click on their links to learn more about them.
- anvi-script-checkm-tree-to-interactive
- anvi-export-locus
- anvi-delete-hmms
- anvi-get-sequences-for-gene-clusters
- anvi-import-misc-data
- anvi-export-misc-data
- anvi-show-misc-data
- anvi-delete-misc-data
- anvi-merge-bins
- anvi-meta-pan-genome
- anvi-migrate-db
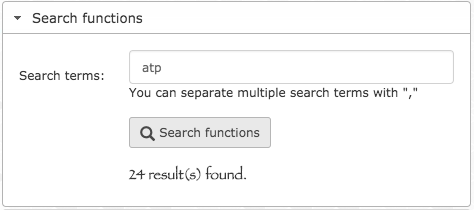
- anvi-search-functions
Thanks
We know that developing anvi'o would have been much less fun without its enthusiastic and engaged users. We are thankful for those, including Bryan Merrill, Rika Anderson, Mike Lee, Marta Royo-Llonch, Xabier Vázquez-Campos, Emily Crossette, Varun Srinivasan, Julie Reveillaud, Alban Mathieu, and others, who help us improve anvi'o with their science, patience, issue reports, and suggestions.
We are also thankful to our users that share their experiences, such as Elaina Graham, who recently wrote about importing GhostKOALA/KEGG annotations into anvi'o, and Bryan Merrill, who shared his experience with importing VirSorter annotations into anvi'o to study phages for making anvi'o more accessible to the community.