Home
- Introduction to proteoQ
- Installation
- Part I — Data normalization
- Part II — Basic informatics
- Part III — Labs
- References
Chemical labeling using tandem mass tag
(TMT) has been commonly
applied in mass spectrometry (MS)-based quantification of proteins and
peptides. The proteoQ tool is designed to aid automated and
reproducible analysis of proteomics data. It interacts with an Excel
spread sheet for dynamic sample selections, aesthetics controls and
statistical modelings. The arrangement allows users to put data
manipulation behind the scene and apply metadata to openly address
biological questions using various informatic tools. In addition, the
entire workflow is documented and can be conveniently reproduced upon
revisiting.
The tool currently processes the peptide spectrum matches (PSM) tables from Mascot searches for 6-, 10- or 11-plex TMT experiments. Peptide and protein results are then produced with users’ selection of parameters in data filtration, alignment and normalization. The package further offers a suite of tools and functionalities in statistics, informatics and data visualization by creating ‘wrappers’ around published R routines.
To install this package, start R (version “3.6”) and enter:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c("Biobase", "Mfuzz", "limma"))
if (!requireNamespace("devtools", quietly = TRUE))
install.packages("devtools")
devtools::install_github("qzhang503/proteoQ")In this section we illustrate the following applications of proteoQ:
- Summarization of PSM data to peptide and protein reports.
- Visualization of quality metrics in peptide and protein data.
- Re-normalization of data in partial or in full
The data set we use in this section corresponds to the proteomics data
from Mertins et al. (2018). In the study, two different breast cancer
subtypes, triple negative (WHIM2) and luminal (WHIM16), from
patient-derived xenograft (PDX) models were assessed by three
independent laboratories. At each site, lysates from WHIM2 and WHIM16
were each split and labeled with 10-plex TMT at equal sample sizes and
repeated on a different day. This results in a total of 60 samples
labeled under six 10-plex TMT experiments. The samples under each
10-plex TMT were fractionated by off-line, high pH reversed-phase
(Hp-RP) chromatography, followed by LC/MS analysis. The raw PSM
results from Mascot searches
are stored in a companion R package, proteoQDA, and are accessbile
through the following installation:
devtools::install_github("qzhang503/proteoQDA")We first set up a working directory:
dat_dir <- "c:\\The\\First\\Example"The workflow begins with PSM table(s) in a csv format from the
Mascot search engine. When
exporting PSM results, I typically set the option of
Include sub-set protein hits to 0 with my opinionated choice in
satisfying the principle of parsimony. The options of Header and
Peptide quantitation should be checked to include the search
parameters and quantitative values, respectively. The file name(s) of
the exports will be taken as is.[1]
The same peptide sequence under different PSM files can be assigned to
different protein IDs when
inferring proteins
from peptides using algorithms such as greedy set cover. To avoid such
ambiguity in protein inference, I typically enable the option of
Merge MS/MS files into single search in Mascot
Daemon. If the option is
disabled, peptide sequences that have been assigned to multiple protein
IDs will be removed for now when constructing peptide reports.
The merged search may become increasingly cumbersome with growing data
sets. In this example, I combined the MS peak lists from the Hp-RP
fractions within the same 10-plex TMT experiment, but not the lists
across experiments. This results in a total of six pieces of PSM results
in Mascot exports. To get us started, we go ahead and copy over the
PSM files that we have prepared in proteoQDA to the working directory:
library(proteoQDA)
cptac_csv_1(dat_dir)The workflow involves an Excel template containing the metadata of
multiplex experiment numbers, including TMT channels, LC/MS injection
indices, sample IDs, reference channels, RAW MS data file names and
addditional fields from the users. The default file name for the
experimental summary is expt_smry.xlsx. If samples were fractionated
off-line prior to LC/MS, a second Excel template will also be filled
out to link multiple RAW MS file names that are associated to the same
sample IDs. The default file name for the fractionation summary is
frac_smry.xlsx.[2] Unless otherwise mentioned, we will assume these
default file names throughtout the document.
Columns in the expt_smry.xlsx are approximately divided into the
following three tiers: (1) essential, (2) optional default and (3)
optional open. We supply the required information of the TMT
experiments under the essential columns. The optional default columns
serve as the fields for convenient lookups in sample selection,
grouping, ordering, aesthetics etc. For instance, the program will by
default look for values under the Color column if no instruction was
given in the color coding of a PCA plot. The optional open fields on the
other hand allow us to define our own analysis and aesthetics: we may
openly define multiple columns of contrasts at different levels of
granularity for uses in linear modeling. Description of the column keys
can be found from the help document by entering ?proteoQ::load_expts
from a R console.
We next copy over a pre-compiled expt_smry.xlsx and a frac_smry.xlsx
to the working directory:
cptac_expt_1(dat_dir)
cptac_frac_1(dat_dir)We now have all the pieces that are required by proteoQ in place.
Let’s have a quick glance at the expt_smry.xlsx file. We note that no
reference channels were indicated under the column Reference. With
proteoQ, the log2FC of each species in a given sample is calculated
either (a) in relative to the reference(s) within each multiplex TMT
experiment or (b) to the mean of all samples in the same experiment if
reference(s) are absent. Hence, the later approach will be employed to
the examplary data set that we are working with. In this special case,
the mean(log2FC) for a given species in each TMT experiment is
averaged from five WHIM2 and five WHIM16 samples, which is
biologically equivalent across TMT experiments.
As a final step of the setup, we will load the experimental summary and some precomputed results:
library(proteoQ)
load_expts()Process PSMs — In this section, we demonstrate the summarisation of
PSM data to peptides and proteins. We start by processing PSM data from
Mascot outputs:
# PSM reports
normPSM(
rptr_intco = 1000,
rm_craps = FALSE,
rm_krts = FALSE,
rm_outliers = FALSE,
plot_violins = TRUE
)
# or accept the default parameters
normPSM()PSM outliers will be assessed at a basis of per peptide and per sample
at rm_outliers = TRUE, which can be a slow process for large data
sets. To circumvent repeated efforts in the assessment of PSM outliers,
we may set rm_outliers = FALSE and plot_violins = TRUE when first
executing normPSM(). We then visually inspect the violin plots of
reporter-ion intensity. Empirically, PSMs with reporter-ion intensity
less than 1,000 are trimmed and samples with median intensity that is
2/3 or less to the average of majority samples are removed from further
analysis.[3]
Summarize PSMs to peptides — We next summarise PSM to peptides.
# peptide reports
normPep(
id = pep_seq,
method_psm_pep = median,
method_align = MGKernel,
range_log2r = c(5, 95),
range_int = c(5, 95),
n_comp = 3,
seed = 749662,
maxit = 200,
epsilon = 1e-05
)At id = pep_seq_mod, peptide sequences that are different in variable
modificaitons will be treated as different species. The log2FC of
peptide data will be aligned by median centering across samples by
default. If method_align = MGKernel is chosen, log2FC will be
aligned under the assumption of multiple Gaussian kernels.[4] The
parameter n_comp defines the number of Gaussian kernels and seed set
a seed for reproducible fittings. The parameters range_log2r and
range_int define the range of log2FC and the range of reporter-ion
intensity, respectively, for use in the scaling of standard deviation
across samples.
Let’s compare the log2FC profiles with and without scaling
normalization:[5]
# without scaling
pepHist(
scale_log2r = FALSE,
ncol = 10
)
# with scaling
pepHist(
scale_log2r = TRUE,
ncol = 10
)By default, the above calls will look for none void entries under column
Select in expt_smry.xlsx. This will results in histogram plots with
60 panels in each, which may not be easy to explore as a whole. In
stead, we will break the plots down by their data origins. We begin with
modifying the expt_smry.xlsx file by adding the columns BI, JHU
and PNNL. Each of the new columns includes sample entries that are
tied to their laboratory origins.
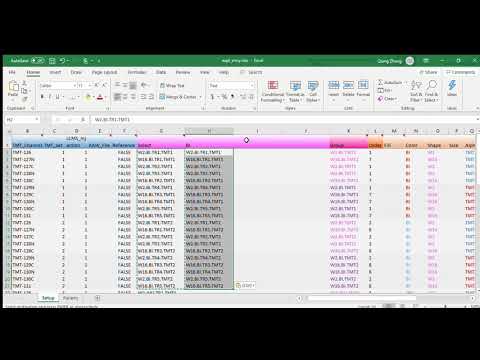
{kind=link}
{kind=link}
{kind=link}
We now are ready to plot histograms for each subset of data.[6] In this
document, we only display the plots using the BI subset:
# without scaling
pepHist(
scale_log2r = FALSE,
col_select = BI,
filename = Hist_BI_N.png,
ncol = 5
)
# with scaling
pepHist(
scale_log2r = TRUE,
col_select = BI,
filename = Hist_BI_Z.png,
ncol = 5
)NB: We interactively told pepHist() that we are interested in sample
entries under the newly created BI column. Behind the scene, the
interactions are facilitated by
openxlsx
via the readind and writing of the Setup workbook in expt_smry.xlsx.
We also supply a file name, assuming that we want to keep the earlierly
generated plots with default file names of Peptide_Histogram_N.png and
Peptide_Histogram_Z.png.


**Figure 1.** Histograms of peptide log2FC. Left: `scale_log2r = FALSE`; right, `scale_log2r = TRUE`
As expected, the widths of log2FC profiles become more consistent
after the scaling normalization. However, such adjustment may cause
artifacts when the standard deviaiton across samples are genuinely
different. I typically test scale_log2r at both TRUE and FALSE,
then make a choice in data scaling together with my a priori knowledge
of the characteristics of both samples and references.[7] We will use
the same data set to illustrate the impacts of reference selections in
scaling normalization in Lab 3.1.
Alignment of log2FC against housekeeping or normalizer protein(s) is
also available. This seems suitable when sometime the quantities of
proteins of interest are different across samples where the assumption
of constitutive expression for the vast majority of proteins may not
hold.
Summarize peptides to proteins — We then summarise peptides to proteins using a two-component Gaussian kernel.[8]
# protein reports
normPrn(
id = gene,
method_pep_prn = median,
method_align = MGKernel,
range_log2r = c(5, 95),
range_int = c(5, 95),
n_comp = 2,
seed = 749662,
fasta = "C:\\Results\\DB\\Refseq\\RefSeq_HM_Frozen_20130727.fasta",
maxit = 200,
epsilon = 1e-05
)Similar to the peptide summary, we inspect the alignment and the scaling of ratio profiles:
# without scaling
prnHist(
scale_log2r = FALSE,
ncol = 10
)
# with scaling
prnHist(
scale_log2r = TRUE,
ncol = 10
)NB: At this point, we might have reach a consensus on the choice of
scaling normalization. If so, it may be plausible to set the value of
scale_log2r under the Global environment, which is typically the R
console that we are interacting with.
# if agree
scale_log2r <- TRUE
# or disagree
scale_logr <- FALSEIn this way, we can skip the repetitive setting of scale_log2r in our
workflow from this point on, and more importantly, prevent ourselves
from peppering the settings of TRUE or FALSE from calls to
calls.
A multi-Gaussian kernel can fail capturing the log2FC profiles for a
subset of samples. This is less an issue with a small number of samples.
Using a trial-and-error approach, we can start over with a new
combination of parameters, such as a different seed, and/or a
different range of scale_log2r et al. However, the one-size-fit-all
attempt may remain inadequate when the number of samples is relatively
large. The proteoQ allow users to focus fit aganist selected
samples. This is the job of argument col_refit. Let’s say we want to
re-fit the log2FC for samples W2.BI.TR2.TMT1 and W2.BI.TR2.TMT2.
We simply add a column, which I named it Select_sub, to
expt_smry.xlsx with the sample entries for re-fit being indicated
under the column:

We then execute the following codes with argument col_fit being linked
to the newly created column:
normPep(
id = pep_seq,
method_psm_pep = median,
method_align = MGKernel,
range_log2r = c(5, 95),
range_int = c(5, 95),
n_comp = 3,
col_refit = Select_sub,
seed = 749662,
maxit = 200,
epsilon = 1e-05,
)In this section I illustrate the following applications of proteoQ:
- Basic informatic analysis against peptide and protein data.
- Linear modeling using contrast fits
We first visualize MDS, PCA and Euclidean distance against the peptide data. We start with metric MDS for peptide data:
# all data sets
pepMDS(
show_ids = FALSE
)
**Figure 2A.** MDS of peptide log2FC at `scale_log2r = TRUE`
It is clear that the WHIM2 and WHIM16 samples are well separated by the
Euclidean distance of log2FC (Figure 2A). We next take the JHU
data subset as an example to explore batch effects in the proteomic
sample handling:
# `JHU` subset
pepMDS(
col_select = JHU,
filename = MDS_JHU.png,
show_ids = FALSE
)

**Figure 2B-2C.** MDS of peptide log2FC for the `JHU` subset. Left: original aesthetics; right, modefied aesthetics
We immediately spot that all samples are coded with the same color
(Figure 2B). This is not a surprise as the values under column
expt_smry.xlsx::Color are exclusively JHU for the JHU subset. For
similar reasons, the two different batches of TMT1 and TMT2 are
distinguished by transparency, which is governed by column
expt_smry.xlsx::Alpha. We may wish to modify the aesthetics using
different keys: e.g., color coding by WHIMs and size coding by batches,
without the recourse of writing new R scripts. One solution is to link
the attributes and sample IDs by creating additional columns in
expt_smry.xlsx. In this example, we have had coincidentally prepared
the column Shape and Alpha to code WHIMs and batches, respectively,
for the JHU subset. Therefore, we can recycle them directly to make a
new plot (Figure 2C):
# `JHU` subset
pepMDS(
col_select = JHU,
col_fill = Shape, # WHIMs
col_size = Alpha, # batches
filename = MDS_JHU_new_aes.png,
show_ids = FALSE
)Accordingly, the prnMDS performs MDS for protein data. For PCA
analysis, the corresponding functions are pepPCA and prnPCA for
peptide and protein data, respectively.
While MDS approximates Euclidean distances at a low dimensional space.
Sometime it may be useful to have an accurate view of the distance
matrix. Functions pepEucDist and prnEucDist plot the heat maps of
Euclidean distance matrix for peptides and proteins, respectively. They
are wrappers of
pheatmap
and inherit many parameters therein. Supposed that we are interested in
visualizing the distance matrix for the JHU subset:
# `JHU` subset
pepEucDist(
col_select = JHU,
annot_cols = c("Shape", "Alpha"),
annot_colnames = c("WHIM", "Batch"),
# parameters from `pheatmap`
display_numbers = TRUE,
number_color = "grey30",
number_format = "%.1f",
clustering_distance_rows = "euclidean",
clustering_distance_cols = "euclidean",
fontsize = 16,
fontsize_row = 20,
fontsize_col = 20,
fontsize_number = 8,
cluster_rows = TRUE,
show_rownames = TRUE,
show_colnames = TRUE,
border_color = "grey60",
cellwidth = 24,
cellheight = 24,
width = 14,
height = 12,
filename = EucDist_JHU.png
)Parameter annot_cols defines the tracks to be displayed on the top of
distrance-matrix plots. In this example, we have choosen
expt_smry.xlsx::Shape and expt_smry.xlsx::Alpha, which encodes the
WHIM subtypes and the batch numbers, respectively. Parameter
annot_colnames allows us to rename the tracks from Shape and Alpha
to WHIM and Batch, respectively, for better intuition. We can
alternatively add columns WHIM and Batch if we choose not to recycle
columns Shape and Alpha.

**Figure 2D.** EucDist of peptide log2FC at `scale_log2r = TRUE`
In this section, we visualize the batch effects through correlation
plots. The proteoQ tool currently limits itself to a maximum of 44
samples for a correlation plot. In the document, we will perform
correlation analysis against the PNNL data subset. By default, samples
will be arranged diagnoally from upper left to bottom right by the row
order of expt_smry.xlsx::Select. We have learned from the earlier
MDS analysis that the batch effects are smaller than the differences
between W2 and W16. We may wish to put the TMT1 and TMT2 groups
adjacient to each other for visualization of more nuance batch effects,
followed by the correlational comparison of WHIM subtypes. We can
achieve this by supervising sample IDs at a customized order. In the
expt_smry.xlsx, I have prepared an Order column where samples within
the JHU subset were arranged in the descending order of W2.TMT1,
W2.TMT2, W16.TMT1 and W16.TMT2. Now we tell the program to look
for the Order column for sample arrangement:
# peptide correlation
pepCorr(
col_select = PNNL,
col_order = Order,
filename = PNNL.png,
use_log10 = TRUE,
scale_log2r = TRUE,
min_int = 3.5,
max_int = 6.5,
min_log2r = -2,
max_log2r = 2,
width = 24,
height = 24
)
# protein correlation
prnCorr(
col_select = PNNL,
col_order = Order,
filename = PNNL.png,
use_log10 = TRUE,
scale_log2r = TRUE,
min_int = 3.5,
max_int = 6.5,
min_log2r = -2,
max_log2r = 2,
width = 24,
height = 24,
)

**Figure 3A-3B.** Correlation of log2FC for the `PNNL` subset. Left: peptide; right, protein
The following performs the imputation of peptide and protein data. More
information can be found from
mice.
# peptides
pepImp(m = 2, maxit = 2)
# proteins
prnImp(m = 5, maxit = 5)The following performs heat map visualization against protein data.
Detailed description of the arguments can be found from
pheatmap
and ?prnHM.
# protein heat maps
prnHM(
xmin = -1,
xmax = 1,
x_margin = 0.1,
annot_cols = c("Group", "Color", "Alpha", "Shape"),
annot_colnames = c("Group", "Lab", "Batch", "WHIM"),
cluster_rows = TRUE,
cutree_rows = 10,
show_rownames = FALSE,
show_colnames = TRUE,
fontsize_row = 3,
cellwidth = 14,
width = 18,
height = 12,
)
**Figure 4.** Heat map visualization of protein log2FC
In this section, we perform the significance analysis of peptide and
protein data. The approach of contrast fit is used here (Chambers, J. M.
(1992) Linear models; limma, Gordon Smith). We first define the
contrast groups for significance tests. For this purpose, I have devided
the samples by their WHIM subtypes, laboratory locations and batch
numbers. This ends up with entries of W2.BI.TMT1, W2.BI.TMT2 etc.
under the expt_smry.xlsx::Term column. The interactive environment
between the Excel file and the proteoQ tool allows us to enter more
columns of contrasts when needed. For instance, we might also be
interested in a more course comparison of inter-laboratory differences
without batch effects. The corresponding contrasts of W2.BI, W2.BI
etc. can be found under a pre-made column, Term_2. Having these
columns in hand, we are now ready to perform significance tests for
peptides and protein species. In the document, we will analyze protein
data and perform volcano plot visualization:
# significance tests of protein log2FC
prnSig(
impute_na = FALSE,
W2_bat = ~ Term["(W2.BI.TMT2-W2.BI.TMT1)", "(W2.JHU.TMT2-W2.JHU.TMT1)", "(W2.PNNL.TMT2-W2.PNNL.TMT1)"], # batch effects
W2_loc = ~ Term_2["W2.BI-W2.JHU", "W2.BI-W2.PNNL", "W2.JHU-W2.PNNL"], # location effects
)
# volcano plots
prnVol()Note that we have informed the prnSig function to look for contrasts
under columns Term and Term_2, followed by the cotrast pairs in
square brackets. Pairs of contrasts are separated by commas.

**Figure 5A.** Volcano plots of protein log2FC between two batches.

**Figure 5B.** Volcano plots of protein log2FC between locations.
There are a handful of R tools for gene set enrichement analysis, such
as GSEA, GSVA, gage, to name a few. It may be intuitive as well if we
can visualize the enrichment of gene sets under the context of volcano
plots. Currently, the preoteoQ takes a naive approach to visualize the
asymmetricity of protein probability p-values under volcano plots.
In the analysis of Gene Set Probability Asymmetricity (GSPA), the
significance pVals of proteins obtained from linear modeling are
taken, followed by the calculation of the geometric mean of pVals for
the groups of up- or down-regulated proteins within a gene set, as well
as the corresponding mean log2FC. The quotient of the two pVals is
then taken to represent the significance of enrichment and the delta of
the two log2FC for use as the fold change of enrichment. The arguments
pval_cutoff and logFC_cutoff allow us to filter out low impact genes
prior to the analysis.
prnGSPA(
impute_na = FALSE,
pval_cutoff = 5E-2,
logFC_cutoff = log2(1.2),
gset_nm = c("go_sets", "kegg_sets"),
)To visualize the distribution of protein log2FC and pVal within gene
sets:
gspaMap(
show_labels = TRUE,
pval_cutoff = 1E-2,
logFC_cutoff = log2(1.2),
show_sig = pVal,
yco = 0.01,
)This will produce the volcano plots of proteins within gene sets that have passed our selection criteria. Here, we show one of the examples:

**Figure 6.** An example of volcano plots of protein log2FC under a gene set
The following performs the trend analysis against protein expressions.
More information can be found from
Mfuzz.
Note that the number of clusters is provided by n_clust, which can be
a single value or a vector of integers.
# soft clustering of protein data
anal_prnTrend(
scale_log2r = TRUE,
n_clust = 6
)
# visualization
plot_prnTrend()
**Figure 7.** Trend analysis of protein log2FC.
The following performs the NMF analysis against protein data. More
details can be found from
NMF.
# load library
library(NMF)
# NMF analysis
anal_prnNMF(
# col_group = Group, # optional a priori knowledge of sample groups
scale_log2r = TRUE,
r = 6,
nrun = 200
)
# consensus heat map
plot_prnNMFCon(
r = 6,
annot_cols = c("Color", "Alpha", "Shape"),
annot_colnames = c("Lab", "Batch", "WHIM"),
width = 10,
height = 10
)
# coefficient heat map
plot_prnNMFCoef(
r = 6,
annot_cols = c("Color", "Alpha", "Shape"),
annot_colnames = c("Lab", "Batch", "WHIM"),
width = 10,
height = 10
)
# metagene heat map(s)
plot_metaNMF(
r = 6,
annot_cols = c("Color", "Alpha", "Shape"),
annot_colnames = c("Lab", "Batch", "WHIM"),
fontsize = 8,
fontsize_col = 5
)

**Figure 8A-8B.** NMF analysis of protein log2FC. Left: concensus; right: coefficients.
In this lab, we explore the effects of reference choices on data
normalization. We first copy data over to the file directory specified
by temp_dir, followed by PSM, peptide normalization and histogram
visualization of peptide log2FC.
# directory setup
temp_dir <- "c:\\The\\W2_ref\\Example"
library(proteoQDA)
cptac_csv_1(temp_dir)
cptac_expt_ref_w2(temp_dir)
cptac_frac_1(temp_dir)
# analysis
library(proteoQ)
load_expts(temp_dir, expt_smry_ref_w2.xlsx)
normPSM()
normPep(
id = pep_seq,
method_psm_pep = median,
method_align = MGKernel,
range_log2r = c(5, 95),
range_int = c(5, 95),
n_comp = 3,
seed = 911,
maxit = 200,
epsilon = 1e-05
)
# visualization
pepHist(
scale_log2r = FALSE,
ncol = 9
)
**Figure S1A.** Histograms of peptide log2FC with a WHIM2 reference.
Notice that in the above histograms the log2FC profiles of WHIM2
samples are much narrower than those of WHIM16 (Figure S1A). This
will occur when a reference is more similar to one group of sample(s)
than the other. In our case, the reference is one of WHIM2. The
difference in the breadth of log2FC profiles between the WHIM16 and
the WHIM2 groups is likely due to the genuine difference in their
proteomes. If the above argument is valid, a scaling normalize would
moderate, and thus bias, the quantitative difference in proteomes
between WHIM2 and WHIM16.
We alternatively seek a “center-of-mass” representation for uses as
references. We select one WHIM2 and one WHIM16 from each 10-plex
TMT. The proteoQ tool will average the signals from designated
references. Thefore, the derived reference can be viewed as a mid point
of the WHIM2 and the WHIM16 proteomes. We next perform analogously
the data summary and histogram visualization. With the new reference, we
have achieved log2FC profiles that are more comparable in breadth
between WHIM2 and WHIM16 samples. With the new reference, a scaling
normalization may be suitable for later steps.
# directory setup
temp_dir_2 <- "c:\\The\\W2_W16_ref\\Example"
library(proteoQDA)
cptac_csv_1(temp_dir_2)
expt_smry_ref_w2_w16(temp_dir_2)
cptac_frac_1(temp_dir_2)
# experiment upload
library(proteoQ)
load_expts(temp_dir_2, expt_smry_ref_w2_w16.xlsx)
# PSM normalization
normPSM()
# peptide normalization
normPep(
id = pep_seq,
method_psm_pep = median,
method_align = MGKernel,
range_log2r = c(5, 95),
range_int = c(5, 95),
n_comp = 3,
seed = 911,
maxit = 200,
epsilon = 1e-05,
)
# visualization
pepHist(
scale_log2r = FALSE,
ncol = 8
)
**Figure S1B.** Histograms of peptide log2FC with a combined WHIM2 and WHIM16 reference.
In addition to the global proteomes, the CPTAC publication contains
phosphopeptide data from the same samples.(2018) In this lab, we will
explore the stoichiometry of phosphopeptide subsets in relative to the
combined data sets of global + phospho peptides. We first performed a
search aganist the combined data. The search results are available in
proteoQDA. We next copy the result files over, followed by the
analysis and visualization of the BI subset:
# directory setup
temp_phospho_dir <- "c:\\The\\Phosphopeptide\\Example"
library(proteoQDA)
cptac_csv_2(temp_phospho_dir)
cptac_expt_2(temp_phospho_dir)
cptac_frac_2(temp_phospho_dir)
# experiment upload
library(proteoQ)
load_expts(temp_phospho_dir, expt_smry.xlsx)
# PSM normalization
normPSM()
# peptide normalization
normPep(
id = pep_seq_mod, # peptides with different variable modifications
method_psm_pep = median,
method_align = MGKernel,
range_log2r = c(5, 95),
range_int = c(5, 95),
n_comp = 3,
seed = 749662,
maxit = 200,
epsilon = 1e-05
)
# histograms for all peptides
pepHist(
col_select = BI,
scale_log2r = TRUE,
ncol = 4,
filename = "BI_all_peptides.png"
)
# histograms for phosphopeptide subsets
pepHist(
col_select = BI,
scale_log2r = TRUE,
pep_pattern = "sty",
ncol = 4,
filename = "BI_pSTY.png"
)

**Figure S2A-S2B.** Histogram visualization of peptide log2FC. Left: global + phospho; right: phospho only.
Ideally, the profiles of the log2FC between the phospho subsets and
the overall data would either align at the maximum density or perhaps
offset by similar distance among replicated samples. In this example,
the alginment at maximum density seems to be case. The observation
raises the possibility of measuring the stoichiometry of
phosphoproteomes in relative to global data across sample types or
conditions.
NB: I used underscore to denote N-terminal acetylation. The R language
will throw an error if we attemp to use pep_pattern = _ to subset
peptides with N-terminal acetylation. In this case, we will need to
quote the underscore: pep_pattern = "_".
Single random effect — In proteomic studies involved multiple
multiplex TMT experiments, the limited multiplicity of isobaric tags
requires sample parting into subgroups. Measures in log2FC are then
obtained within each subgroup by comparing to common reference
materials, followed by data bridging across experiments. This setup
violates the independence assumption in statistical sampling as the
measures of log2FC are batched by TMT experiments. In this lab, we
will use the CPTAC data to test the statistical significance in protein
abundance between the WHIM2 and the WHIM16 subtypes, by first taking
the batch effects into account. We will use mixed-effects models to
explore the random effects that were introduced by the data stitching.
In case that you would like to find out more about mixed-effects models
in R, I found the online
tutorial a
helpful resource.
We start off by copying over the expt_smry.xlsx file, which contains a
newly created column, Term_3, for terms to be used in the statistical
tests of WHIM2 and WHIM16. We also copy over the protein results
from Part I of the vignette and carry out the signficance tests with
and without random effects.
# directory setup
temp_raneff_dir <- "c:\\The\\Random_effects\\Example"
library(proteoQDA)
cptac_prn_1(temp_raneff_dir)
cptac_expt_3(temp_raneff_dir)
cptac_frac_3(temp_raneff_dir)
# analysis
library(proteoQ)
load_expts(temp_raneff_dir, expt_smry.xlsx)
prnSig(
impute_na = FALSE,
W2_vs_W16_fix = ~ Term_3["W16-W2"], # fixed effect only
W2_vs_W16_mix = ~ Term_3["W16-W2"] + (1|TMT_Set), # one fixed and one random effects
)
# volcano plots
prnVol()In the formula linked to argument W2_vs_W16_mix, the random effect
(1|TMT_Set) is an addition to the fix effect Term_3["W16-W2"]. The
syntax (1|TMT_Set) indicates the TMT_Set term to be parsed as a
random effect. The name of the term is again a column key in
expt_smry.xlsx. In this example, the TMT batches are documented
under the column TMT_Set and can be applied directly to our formula.
Upon the completion of the protein signficance tests, we can analyze
analogously the gene set enrichment against these new formulas by
calling functions prnGSPA and gspaMAP.
Multiple random effects — In this section, we will test the
statistical significance in protein abundance changes between the
WHIM2 and the WHIM16 subtypes, by taking additively both the TMT
batch effects and the laboratory effects into account. At the time of
writing the document, I don’t yet know how to handle multiple random
effects using limma. Alternatively, I use lmerTest to do the work.
Missing values can frequently fail random-effects modeling with more
complex error structures and need additional cares. One workaround is to
simply restrict ourselves to entries that are complete in cases. This
would lead to a number of proteins not measurable in their statistical
significance. Alternatively, we may seek to fill in missing values using
techniques such as multivariate imputation. At present, I use the mice
function from the R package “mice” in prnImp() and pepImp() for the
imputation of protein and peptide data, respectively. To impute protein
data:
prnImp(m = 5, maxit = 5)We further note that the laboratory differences are coded under columns
Color in expt_smry.xlsx. We then test the statistical difference
between WHIM2 and WHIM16 aganist the following three models:
prnSig(
impute_na = TRUE,
method = lm,
W2_vs_W16_fix = ~ Term_3["W16-W2"], # one fixed effect
W2_vs_W16_mix = ~ Term_3["W16-W2"] + (1|TMT_Set), # one fixed and one random effect
W2_vs_W16_mix_2 = ~ Term_3["W16-W2"] + (1|TMT_Set) + (1|Color), # one fixed and two random effects
)
# correlation plots
read.csv(file.path(temp_raneff_dir, "Protein\\Model\\Protein_pVals.txt"),
check.names = FALSE, header = TRUE, sep = "\t") %>%
dplyr::select(grep("pVal\\s+", names(.))) %>%
`colnames<-`(c("none", "one", "two")) %>%
dplyr::mutate_all(~ -log10(.x)) %>%
GGally::ggpairs(columnLabels = as.character(names(.)), labeller = label_wrap_gen(10), title = "",
xlab = expression("pVal ("*-log[10]*")"), ylab = expression("pVal ("*-log[10]*")")) The correlation plots indicate that the random effects of batches and
laboratory locations are much smaller than the fixed effect of the
biological differences of WHIM2 and WHIM16.

**Figure S3.** Pearson r of protein significance p-values.
Philipp, Martins. 2018. “Reproducible Workflow for Multiplexed Deep-Scale Proteome and Phosphoproteome Analysis of Tumor Tissues by Liquid Chromatography-Mass Spectrometry.” Nature Protocols 13 (7): 1632–61. https://doi.org/10.1038/s41596-018-0006-9.
[1] The default file names begin with letter F, followed by six digits
and ends with .csv in name extension.
[2] To extract the names of RAW files under a raw_dir folder:
extract_raws(raw_dir)
[3] The sample removal and PSM re-processing can be achieved by deleting
the corresponding entries under the column Sample_ID in
expt_smry.xlsx, followed by the re-load of the experiment,
load_expts(), and the re-execution of normPSM() with desired
parameters.
[4] Density kernel estimates can occasionally capture spikes in the profiles of log2FC during data alignment. Users will need to inspect the alignment of ratio histograms and may optimize the data normalization in full with different combinations of tuning parameters or in part against a subset of samples, before proceeding to the next steps.
[5] normPep() will report log2FC results both before and after the
scaling of standard deviations.
[6] System files will be automatically updated from the modified
expt_smry.xlsx
[7] The default is scale_log2r = TRUE throughout the package. When
calling functions involved parameter scale_log2r, users can specify
explicitly scale_log2r = FALSE or define its value under the global
environment.
[8] Prameter fasta is solely used for the calculation of protein
percent coverage. Precomputed data will be used if no fasta database
is provided.